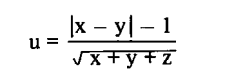
6. MATERIALS AND METHODS
6. 1. Organisms
Several strains were isolated from the inner part of young fruit bodies (Agaricus bisporus, strain A1; Agaricus bisporus, strain 18; Agaricus bitorquis; Calvatia gigantea; Coprinus comatus, strain L). One strain, namely that of Armillaria mellea, was isolated from spores. Other strains were obtained from the CBS, IFO and NRRL culture collections (Centraal Bureau voor Schimmelcultures, Baarn, the Netherlands; Institute for Fermentation, Osaka, Japan; and Northern Regional Research Laboratory, Peoria, Ill., U.S.A., respectively).
For investigation of the flavour fresh fruit bodies of Agaricus bisporus and Lactarius sanguifluus were obtained commercially. Fruit bodies of Agaricus bitorquis, Calvatia gigantea, Coprinus comatus, Pholiota squarrosa and Tricholoma nudum were collected in the field. Fruit bodies of Pleurotus ostreatus were grown on sterilised sawdust supplemented with malt extract and casein. This medium was inoculated with a mycelial culture of the fungus (NRRL 2366). Canned Boletus edulis, Cantharellus cibarius and Tricholoma portentosum, dried Boletus edulis, Gyromitra esculenta and Marasmius scorodonius, as well as fresh potatoes and tomatoes, were purchased commercially. Dried Lentinus edodes was kindly furnished by Dr. N. Hiratsuka (Chofu-shi, Tokyo, Japan).
6.2. Media
For investigation of the nutritional requirements the basal medium contained per litre deionized water 30 g (for Agaricus bisporus) or 60 g (for Coprinus comatus) glucose monohydrate, 16 g casein, 2 g K2HP04, 0.4 g MgS04.7 H20, 0.2 g NaCl, 0.02 g CaCl2.2 H20, 10 mg FeCl3.6 H20, 7.2 mg MnCl2.4 H20, 4 mg ZnCl2, 1.25 Mg CuS04.5 H20, 0.4 mg thiamine hydrochloride and 50 mg adenine (the last mentioned substance only for Coprinus comatus).
The influence of the carbon and energy source was studied by replacing glucose with other compounds. The complete media were sterilised at 110 °C for 20 min. To investigate the effect of Maillard compounds on the growth every carbohydrate was sterilised separately and added aseptically to the sterile basal medium after cooling. In this procedure the carbohydrate and the rest of the medium were each dissolved in half the amount of water. In some experiments the saccharose was hydrolysed by boiling with 0.01 N HCl for 5 min followed by neutralisation with KOH. Volatile carbon sources (non-sterilised alcohols and hydrocarbons sterilised by filtration) were also added after heat sterilisation of the basal medium. All other carbon sources (lipids, salts of organic acids) were added before heat sterilisation. When saturated lipids were used as carbon source, the culture flasks were incubated on a shaker at 25 °C immediately after sterilisation, so that the hot medium cooled during shaking, resulting in a suspension of small globules of the lipids concerned. The unsaturated lipids emulsified during the first 24 h of shaking.
The influence of the nitrogen source was investigated by replacing casein with other nitrogen compounds. For testing the influence of growth substances thiamine hydrochloride and adenine were omitted from the basal medium and, instead of casein, a mixture of amino acids was used as nitrogen source for Agaricus bisporus and asparagine and phenylalanine for Coprinus comatus (concentrations, see Tables 10 and 11). The growth substances were sterilised by filtration and added aseptically to the heat-sterilised basal medium. A mixture of 8 vitamins was used so that the medium contained per litre 2 μg biotin, 400 mg calcium pantothenate, 2 mg m-inositol, 400 μg niacin, 200 μg p-aminobenzoic acid, 400 μg pyridoxine hydrochloride, 200 μg riboflavine and 400 μg thiamine hydrochloride. A mixture of 4 nucleobases was used, the final medium containing 12.5 mg adenine sulphate, 12.5 mg guanine hydrochloride, 12.5 mg uracil and 12.5 mg xanthine.
The following natural media and ingredients of media were used. Malt extract was prepared from fresh malt. The extract contained approximately 110 g reducing sugars, 160 g total sugars and 1.1 g nitrogen per litre. Casein ("vitamin free") was obtained from Hoffmann-La Roche (Baslé, Switzerland). For fermentor experiments and solid media a less pure casein from Baker Chemicals (Deventer, the Netherlands) was used. CME (casein-malt extract) contained 5 g casein, 250 ml malt extract and 2 g K2HP04 per litre. For Coprinus comatus, Morchella esculenta and Volvariella volvacea the concentration of malt extract was 500 ml/l, because these species grew better with this concentration. Skim milk powder, yeast extract and meat extract were from Oxoid (London). Technical maltose was obtained from Pfanstiehl Laboratories (Waukegan, Ill., U.S.A.). A vegetable extract was prepared from cauliflower leaves and contained 7.8 g total solids, 1.7 g glucose, 3.5 g total sugars and 0.65 g nitrogen per litre. The vegetable oil was a commercial salad oil.
Proflo (cottonseed flour) originated from Traders Protein Division (Fort Worth, Texas, U.S.A.). Corn steep liquor was the usual one applied in fermentation industries. Single cell protein was prepared from the yeast Pichia pastoris, grown in submerged culture with ammonium chloride and methanol as sources of nitrogen and carbon. A concentrated cell suspension of this yeast was incubated at 50 °C during 24 h for autolysis. After centrifugation the clear supernatant was added to a culture medium as nitrogen source.
The following solid media were used: CMEA (casein-malt extract-agar) contained 5 g casein, 250 ml malt extract, 2 g K2HP04 and 10 g agar per litre; asparagine agar contained 10 g glucose, 2 g K2HP04, 0.4 g MgS04. 7 H20, 0.2 g NaCl, 0.02 g CaCl2.2 H20, 5 g asparagine, a mixture of 8 vitamins of the same composition as mentioned before, trace elements (Fe, Mn, Zn and Cu supplied in the same form as in the basal medium) and 20 g Difco agar per litre; vitamin-free asparagine agar had the same composition, with omission of the vitamin mixture.
6.3. Chemicals
For synthetic and semi-synthetic (see note *) media analytical grade chemicals were used. Linoleic acid, linolenic acid, methyl linolenate, methyl stearate and methyl palmitate (grade: puriss.) were obtained from Fluka (Buchs, Switzerland), as were methyl oleate, ethyl oleate, methyl linoleate and ethyl linoleate (grade: purum). Oleic acid (lab. grade) originated from Baker Chemicals (Deventer, the Netherlands), stearic acid (grade: puriss.) from Merck (Darmstadt, B.R.D.) and palmitic acid (purity more than 99%) from BDH Chemicals (Poole, U.K.).
As far as they were commercially available, we used analytical grade chemicals for the organoleptic tests.
The optical isomers of (±)-1 -octen-3-ol were separated by recrystallisation of the strychnine salt of (±)-1-octen-3-ol-hydrogen phthalate, as described by Levene and Walti [92]. The optical isomers of (±)-3-octanol were separated via the brucine salt of the phthalic acid half ester [122].
2-Methyl-2-penten-4-olide was synthesised from mesityl oxide which was first oxidised to acetylmethacrylic acid [34]. The acid did not crystallise but formed an oily fraction. It was reduced with potassium borohydride in a mixture of methanol and water (I/3) at pH 10. After decomposition of the reagent with an excess of hydrochloric acid, 2-methyl-4-hydroxy-2-pentenoic acid was extracted with ether. The ether phase was dried with sodium sulphate and concentrated under reduced pressure. Vacuum distillation yielded a fraction boiling at 50 to 65 °C, showing two major peaks in a gas chromatogram. The compounds were separated with a preparative gas chromatograph on a Carbowax 20 M column, and the second one was found to be 2-methyl-2-penten-4olide. The same lactone has been synthesised from isomesityl oxide by Volger et al. [149]. So far we did not succeed in synthesising an optically active form of this compound as was previously found in Coprinus comatus (Fig. 9A).
1-Octen-3-one was prepared from (±)-1-octen-3-ol by oxidation with potassium bichromate [18] and purified by preparative gas chromatography on a Carbowax 20 M column.
6.4. Culture conditions
Agar cultures. Stock cultures (slants) were maintained in culture tubes containing 8 ml CMEA and were subcultured every two weeks. Petri dishes were inoculated in a sterile room and wrapped in cellophane to prevent contamination during incubation. The incubation time was 2 to 4 weeks. Inocula from agar cultures were used for the liquid media in shaken flasks. The inoculum was taken from asparagine agar for cultures in defined media, from vitamin-free asparagine agar for vitamin studies and from CMEA for complex media. Asparagine agar and vitamin-free asparagine agar were used to reduce the amount of amino acids and growth substances introduced with the inoculum.
Submerged cultures. We prepared inocula for the liquid media in shaken flasks in a sterile room by cutting a colony (diameter about 4 em) from a petri dish and transferring it to a sterile Waring blender with 100 ml water. The colony was homogenised for 1 minute at 18,000 rev/min. The dry weight of mycelium in this suspension was about 120 mg/1. For nutritional studies 100 ml flasks containing 20 ml medium were inoculated with 1 ml of the suspension. The 100 ml flasks were incubated on a purpose-built rotatory shaker (350 rev/min, amplitude 1 cm). Cultures for inoculation of fermentors were made in 500 ml flasks containing 200 ml medium, inoculated with 20 ml suspension of mycelium, and incubated on a Gallenkamp rotatory shaker (180 rev/min, amplitude 2 em).
Fermentor experiments. A Chemap laboratory fermentor containing 10 litres of medium was used. Air was blown in from the bottom and the medium was stirred by a vortex system. The speed of aeration was varied from 1.7 to 6.8 1/min, the stirring rate from 225 to 900 rev/min. To prevent evaporation of the medium, the air was saturated with water. The air-sterilisation filter was heated gently by a heating cord to avoid condensation of water in the filter. The oxygen concentration in the medium and the pH were measured continuously by electrodes. Polypropylene glycol 2000 (0.2%) was used as antifoaming agent. If necessary a few ml of a silicone antifoam emulsion were added during the fermentation.
Unless stated otherwise, the cultures of Agaricus bisporus were incubated for 21 days and those of Coprinus comatus for 14 days; the temperature was kept constant at 250 °C and the initial pH of the media was 7.0.
6.5. Preparation of mushroom extracts
For an organoleptic and chemical investigation of the mushroom flavour aqueous extracts were prepared. One part of dried mushrooms was rehydrated with 23 parts of water, soaked for one hour and homogenised in a Waring blender for 3 min at 18,000 rev/min. The resulting slurry was heated to boiling, kept at 50 °C for one hour and filtered. The clear extract was used for analysis. One part of fresh or canned mushrooms was homogenised with one part of water in a Waring blender and further treated in the same way as the dried mushrooms. Because most fresh mushrooms contain about 8% of dry matter, the two procedures produced extracts of comparable strength. To examine the effect of sterilisation and drying, an amount of cultivated mushrooms (Agaricus bisporus) was divided into three parts before preparation of the aqueous extract. One part was used fresh, another part was first cut into slices and dried at 500 °C to constant weight; the third part was first sterilised at 110 °C for 20 min. Fresh potatoes and tomatoes were treated as fresh mushrooms.
An extract of Agaricus bisporus (200 ml) was evaporated to dryness in a distillation apparatus at 50 °C under reduced pressure, which lasted 4 h. Volatiles were trapped after successive cooling with tap water, ice and liquid nitrogen. After completion of the distillation dry air was blown through the distillation flask into the cooling traps to collect all volatiles remaining in the apparatus.
6.6. Analytical procedures
6.6.1. Chemical and physical methods
Dry weight of mycelium was determined on weighed filter papers or in centrifuge tubes. The culture liquid was removed by filtration or centrifugation. The mycelium was washed three times with water and dried during 48 h at 65 °C or 16 h at 105 °C.
Reducing sugars were measured by the method of Lane and Eynon [119]. Total sugars were established by the anthrone method [108] or with the orcinol reagent [44]. Total nitrogen was determined by the micro-Kjeldahl method [119]. Amino nitrogen was estimated with a quantitative ninhydrin method [106]. Total carbon was analysed with a wet combustion method [15]. The rough lipid content of mycelium was determined gravimetrically after ether extraction in a Soxhlet apparatus for 24 h.
Glucose, fructose, maltose, lactose, galactose, saccharose, L-glutamic acid, L-aspartic acid, urea, 5'-AMP, 5'-ADP and 5'-ATP were determined enzymatically according to procedures described by the manufacturer of the enzymes (Boehringer, Mannheim). The concentrations of 5'-AMP and 5'-ADP could only be ascertained by this method with mushroom extracts. In culture media, after growth of mushroom mycelium, the method revealed high results, contradictory to those of ion exchange experiments, thereby suggesting that substances interfering with the enzymatic reaction were present in the culture media.
Trehalose was measured by ion exchange chromatography [44].
For the determination of laccase a qualitative method devised by Lyr [98] was adapted for quantitative work. After removal of the mycelium by centrifugation at 300 rev/min the culture liquid was diluted 1: 100 with water. Two ml of the solution thus obtained were added to a cuvette (1 cm) containing 1 ml of benzidine reagent. The absorption at 607 nm was measured as a function of time. The increase in the absorption during the first minute (x 10) was arbitrarily defined as the number of units of laccase activity. The benzidine reagent contained 1 g benzidine dissolved in 1000 ml acetate buffer (obtained by mixing 630 ml 0.2 M acetic acid and 370 ml 0.2 M sodium acetate, pH 4.4). The reaction was carried out at 250 °C.
Pellet diameters were measured on enlarged photos of a pellet suspension in a petri dish. The average diameter of 40 to 100 pellets was calculated.
Benzaldehyde, benzyl alcohol, 1-octen-3-ol and 1-octen-3-one were determined in culture liquids, suspensions of mycelium and aqueous mushroom extracts by extracting 8 parts with one part of n-hexane. The hexane phase was analysed with a gas chromatograph, equipped with a 24' x 1/8" stainless steel column with Carbowax 1540 as stationary phase or a 5 m x 3 mm stainless steel column packed with Carbowax 20 M on Chromosorb W-AW. The gas chromatograph operated isothermally at a suitable temperature between 80 and 120 °C. Nitrogen was used as the carrier gas. Acetaldehyde, acetone, ethanol and ethyl acetate were analysed in aqueous mushroom extracts by injecting the extract directly into the gas chromatograph, using the Carbowax 1540 column. The presence of water caused some badly reproducible peaks and disturbed the base line of the chromatograms, so that the detection limit in aqueous samples was higher than in hexane samples.
Isobutyric, isovaleric and n-butyric acids were determined by extracting 8 parts of acidified mushroom extract with one part of diethyl ether which had been freshly double distilled to remove a trace component interfering the analysis of isobutyric acid. The ether phase was analysed with a gas chromatograph, using a 5 m x 3.5 mm glass column packed with terephthalic acid terminated Carbowax 20 M on Chromosorb W-AW. Compounds were quantified with a standard curve, obtained by extracting and analysing aqueous solutions of known compositions.
In most of our GLC-analyses the identification was based on retention data. If necessary, the identification was supported by other techniques, which are specified in the respective tables and the captions of the figures. In these cases the volatiles had to be concentrated. Aqueous mushroom extract or suspensions of mycelium (500 ml) were extracted with 2 x 250 ml isopentane. The isopentane extract was dried with sodium sulphate and the solvent was evaporated. The resulting concentrates had the same smell as the original material. A blank extract was made in the same way starting with 500 ml water. Two μl of the concentrate were injected into the gas chromatograph and the oven temperature was programmed from 70 ° to 150 °C (2° per min). To identify the peaks 10 to 20 μl were injected into a gas chromatograph coupled with a mass spectrometer. For further information some fractions were trapped with liquid nitrogen and analysed with IR, NMR and ORD methods.
Some identifications of alcohols, aldehydes and ketones were supported by oxidation and reduction reactions. Some drops of an aroma concentrate were dissolved in 1 ml of diethyl ether in a test tube. After addition of 1 ml of an aqueous solution, containing 10% potassium bichromate and 15% sulphuric acid, the tube was closed and agitated during 15 min. The ether phase was removed, concentrated and investigated by gas chromatography. The peaks originally representing the alcohols showed a shift to lower retention times in accordance with the expected aldehydes or ketones. This rapid oxidation on a micro scale is a modification of the method of Brown and Garg [18]. Aldehydes and ketones were reduced with sodium borohydride. Some drops of a 10% solution of the reagent in methanol were added to some drops of an aroma concentrate. After 30 min methanol was evaporated and the residue was examined gas chromatographically. The aldehydes and ketones had been reduced to the expected alcohols.
Free amino acids in mushroom extracts and culture liquids were determined with an amino acid analyser using sodium citrate buffers, without preceding purification of the amino acid fraction. The sum of free and bound amino acids in culture liquids and mushroom mycelium was determined after acid hydrolysis (4 h at 145 °C in 6 N HCI). The concentrations of bound amino acids were calculated by subtracting the values for the free amino acids from the values for the respective sums of free and bound amino acids.
Nucleotides and related compounds (elsewhere in this thesis referred to as "nucleotides") were determined in a sample of 80 ml by ion exchange chromatography on Dowex 1-X8 in the formate form. The usual extraction procedure and chromatography [9, 84] were applied without purification of the extract on an acid resin, because this step was not found necessary in our experiments. Peaks in the chromatograms were identified by comparison of the retention times and UV-spectra of the fractions with those of reference compounds and on the basis of results of thin layer chromatography on cellulose-F in the following solvents:
96% ethanol / 1 M ammonium acetate (75/30), pH 7.5 or 3.5;
n-propanol / 12.8 M ammonia water (60/30/10);
saturated ammonium sulphate 1 M sodium acetate / isopropanol (80/20/2);
isobutyric acid / 1 M ammonia (100/60); and on cellulose-PEI (solvent 1 M lithium chloride).
The following reference compounds were available: 5'-ADP; 2',3'-AMP; 5'-AMP; 5'-ATP; 2',3'-CMP; 5'-CMP; 5'-CTP; 5'-GDP; 2',3'GMP; 5'-GMP; 5'-GTP; 5'-IDP; 5'-IMP; 5'-ITP; 5'-UDP; 5'-UDPAG; 2',3'-UMP; 5'-UMP; 5'-UTP; 5'-XDP; 5'-XMP; 5'-XTP. Some fractions not identical with one of these compounds were investigated with other methods. The same extraction procedure and chromatography were applied to 10 litres of culture liquid. The unknown fractions were rechromatographed on the same resin and purified on a Sephadex G 10 column, eluted with water. The purity was checked with thin layer chromatography and the substances were investigated by IR, UV and mass spectrometry.
The flavour threshold concentration was defined as the concentration of a compound in water enabling 50% of the tasters to distinguish this solution from a blank sample. Flavour intensity was expressed in flavour units as proposed by Guadagi et al. [58]. The concentration of a compound or a mixture divided by its threshold concentration was defined as the number of flavour units contributed by that compound or mixture.
Threshold concentrations were determined with an intensity response method [64] using a taste panel of 24 members. For this method the threshold concentration must first be estimated roughly. This was done by a small group of two or three tasters. In some cases only this rough approximation was done. Although no confidence limits can be calculated with this small number of tasters, anyhow somewhat may be said about the accuracy of the rough method. In 14 experiments, where the rough method was followed by the intensity-response method with the whole panel, the results obtained by means of the two methods did never differ by more than a factor of 2.5. Where no confidence limits are given in Chapter 4, the flavour threshold concentrations concerned have been estimated by the rough method.
Differences between samples were examined by presenting pairs of samples (marked A and B) to the tasters and asking them to select the sample of higher flavour intensity or to indicate "no difference".
If brown samples had to be compared with colourless samples, the experiment was performed in almost complete darkness. After the first sessions of the panel, the results of smokers were considered separately; no difference in sensitivity to the flavours investigated was found. No smoking was allowed during tasting.
6.6.3. Statistical interpretation of results
Most nutritional experiments were carried out in four-fold and the results were presented as the average of the four parallels. From 48 observations with Agaricus bisporus and 71 observations with Coprinus comatus the standard deviations in the yields were calculated and found to be 17.6% and 13.1% respectively. Based on these values, the least significant difference (LSD) for comparing averages of four observations was calculated at 95% significance [70]. The high standard deviations were not reduced when the 100 ml flasks were incubated on Gallenkamp shakers instead of the purpose-built shakers. Therefore, to confirm the conclusions, many culture experiments had to be repeated but even then several small differences remained uncertain.
Confidence limits and significance of differences in the determination of flavour threshold concentrations were calculated by the graphic method of Litchfield and Wilcoxon [96].
Results of flavour difference tests were evaluated using the theory of binomial distribution. If results are only influence by change (H0), the eccentricity of a result can be calculated with the formula
where x = the number of judgements "A stronger"
y = the number of judgements "B stronger"
z = the number of judgements "no difference"
The probability (P) of exceeding an eccentricity was taken from the table of the normal distribution. If P was less than 5%, H0 was rejected and the result was considered significant. If P was less than 1%, the result was considered highly significant.
6.6.4. Temperature sensitivity of the Maillard reaction
Culture tubes containing 1 M solutions, pH 6.8, of glucose and glycine, respectively, were sterilised separately. Then the solutions were pooled; the tubes were sealed and incubated at different temperatures (25°, 30°, 37°, 44° and 50 °C). Tubes with the solutions of only glucose or only glycine were also sealed and incubated at the same temperatures. After different time intervals the absorption at 570 nm of the solutions was measured without opening the tubes. The average values for the absorption of the separate solutions of glucose and glycine were subtracted from the corresponding values for the absorption of the pooled solutions incubated at the same temperature. The resulting differences were considered as a measure of the amount of Maillard products.
6.6.5. Fractionation of laccase
Mycelium of Agaricus bisporus was removed from the culture broth by filtration, and laccase was precipitated by adding acetone to a concentration of 60% (vol/vol). The precipitate was centrifuged (6000 rev/min), dissolved in water and dialysed; the resulting solution was then concentrated. The proteins were fractionated on a Sephadex G 200 column, eluted with water. As regards determination of the laccase activity we refer to Item 6.6.1.
Note: *Media with vitamin free casein as the only natural component are termed "semi-synthetic".